Roger B. Cohen, MD, Professor of Medicine and Associate Director of Clinical Research, Abramson Cancer Center, considers the challenges for practicing oncologists in their efforts to interpret and make the best use of the information arising from genomic testing for their patients with cancer. A practicing oncologist himself, Dr. Cohen examines the molecular complexity of common cancers and the many factors, including acquired resistance to therapy, that can confound treatment. Finally, Dr. Cohen reviews the difficulties and deficiencies inherent in the clinical trial system for cancer research.
Related Links
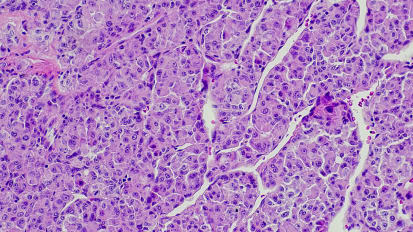
[MUSIC PLAYING] DR. ROGER B. COHEN: This is going to be something completely different. I am trying to put myself in the mindset of a doctor in practice who is trying to cope with the information flow. I'm also a doctor in practice, and I'm trying to cope with the information flow. And it's a little bit overwhelming. So I want to talk about how this might work or how might this work. And I want to talk about not only biological challenges but also cultural challenges. So, I saw this quote a couple of days ago and I liked it. It's from somebody I respect greatly, and he's completely right. There's going to be a lot of this, and we may not like what we find, and we also may not understand it. I think that the complexity is beginning to border on overwhelming. So this is just a slide taken from the literature. It's an outdated slide from 2013. It just shows a bunch of different diseases. The pie charts keep getting more and more complicated. And if you dial in acquired resistance to signaling inhibitors, which is very important and is the subject of biopsies that are being done on patients who are not progressing or responding appropriately to our therapies, you get even more complexity. And so I just highlighted the EGFR inhibitor-acquired mutations, and this is a partial list. The list will undoubtedly continue to grow. Some of these patients will have more than one of these mutations, and so we are learning that molecular complexity is the norm no matter where you look. So I was asked to talk about genomics in clinical trials. Now, we do a pretty lousy job with clinical trials already. So how are we going to do a better job with molecular therapeutics? So we enroll about 3% of patients on clinical trials. We have a hopelessly outdated system for conducting clinical trials, whether you look at trials that are done by pharma or the government. We continue to test new therapies in individual trials. We have more not less review bodies each year. It takes about 36 administrative approvals to get a single Phase 3 trial approved, and it can take about two years. So this clearly is not going to work. And the complexity that's introduced by molecular therapeutics is actually beyond overwhelming, because no individual practice could possibly open or run enough Phase 2 trials to match all the genotypes that are emerging. Nobody can open Phase 2 trials for 1% diseases, not even Memorial Sloan Kettering. And the cost of opening and keeping a trial active for one patient a year is about $10,000 or possibly more, so this is economically not sustainable. So we're going to need a different clinical trials model. Rare mutations are rare, and so these uncommon and rare mutations are going to make it very difficult to do studies to get drugs approved with the existing models. And non-randomized trials, which David Solid alluded to, are probably going to work. And I think the regulators are going to wrap their heads around the idea that if the response rates are spectacular, like ibrutinib, you're done. But they usually will not be quite that good. And randomized trials versus standard of care chemotherapy, I can't think of anything I would rather do less as a practitioner, as a clinical trialist, as a parent, as a husband or a son. These randomized trials are first of all going to be logistically difficult because of the small size of the patients and populations, and they're all over the place, but they're also deeply unappealing to patients and their physicians. So here's a tour de force. So, the French have their act together. They did the Biomarker France project. They showed this at ASCO last year. I was stunned. They can identify and profile all of their non-small-cell lung cancer patients. That's amazing. 10,000 patients, six markers, they find mutations in about half, and then they do a trial. And so they can actually do a trial on 1% of all the lung cancer patients in France who have a BRAF V600E mutation. It's a single arm, Phase 2, dabrafenib. They get a response. 20 patients that were evaluable, and they show that this therapy kind of sort of works a little bit in non-small-cell lung cancer. The problem is France is about the size of Texas, and France has 62 million citizens in a space the size of Texas, and we have 330 million citizens spread over a huge land mass. How are we going to find all these patients, and how are we going to match them to clinical trials? So, this is the information flow as it exists today. And I encourage all of you in the question and answer session to share your own stories. But this is what I do in my practice. We remember who has what mutation. It's on a piece of paper somewhere. I got an email from pathology or foundation. I know I've got it somewhere in my inbox. I'll look for it. I printed a copy for EPIC. My AA was supposed to scan it in. I always copy the result manually into my progress notes. So how should we store all of these data? The in-house generated and the third-party external data, how do we make it accessible and searchable? So, I was talking to David Solid during dinner. And he told me-- I was profoundly reassured by this. He said it is only within the past month or two at Sloan Kettering that they can actually push a button and ask, how many of X do we actually have who are alive and reside within a certain distance from the center? That is a heroic achievement in the current age and something we need to learn how to do. It's complicated. So Dr. [INAUDIBLE] has talked about these variants of undetermined significance, which are growing astronomically. And they're uncertain now, but they may become significant in the future. What are we going to do about them? Who are we going to notify? Do you want to get updated emails every time a variant of undetermined significance becomes significant and then have to notify all of your patients? And then we have this whole issue of tumor heterogeneity and the fact that each patient is a narrative of mutations and genetic changes. So we have to keep track of all of this. So right now we're doing it in our heads. So we know that so and so started off with an EGFR mutation, and then they progressed, and then they got a T790M, and then they progressed, and then they got another mutation. We all remember that narrative in our heads in the individual patient, but that's not going to be tenable in the future. We need a way of keeping up with the patient's updated current molecular status. We need to understand where we're going to keep this information and in what format it's going to be. And genomic reports are at the very best a work in progress. So this is amazing, and it's going to be particularly amazing to the Facebook generation. So this is the information age, and we're using fax technology. And this is what the patients say. I don't know what this means. I just need to study for my mutation. I hope it can be close to my home. Or the patient comes in and says, they sequenced my tumor. They gave me this memory stick and told me to give it to you. Your administrative assistant says, don't worry. I scanned the report into EPIC. It's got to be in there somewhere. And finally the doctor says, please just tell me what to do. So here's a thought problem. This is a real problem. I tried to do this the other day for a patient. I dare you to try searching for a second-generation ALK inhibitor-- pick the one you want-- for a non-small-cell patient with a ROS-1 mutation who's failed crizotinib. This is basic stuff. If you want to make it complicated, add in treated brain mets, stable for eight weeks. And this leads you to conclude that patient-trial matching is not only difficult. It's impossible. And existing matching systems are awful. They don't work. So humans, MDs consistently out perform them, because we have eligibility and eligibility criteria that are too complicated. Many of them are actually pointless, and they're atavisms left over from an earlier age of clinical trials. And we also now have to begin to account for other mutations that could render a patient eligible or ineligible. So we're having a hard enough time just doing it for one like ROS-1, let alone dialing in parallel pathway mutations that a patient might have. So this brings us to basket trials, and Dr. Solid touched on this. There are variations on the theme here. He touched on two of them. I'm going to actually touch on three of them. One of them is reasonably simple, and he talked about it, one mutation, one drug, multiple diseases. I get that. More complicated is multiple mutations, multiple drugs, one disease. I get that. Like lung cancer. And then there's massively complicated, which of course the government has taken on with the so-called MATCH trial, multiple mutations, multiple drugs, multiple diseases, factorial combinations of diseases and patients. So this is the simple one. You've got one mutation like BRAF. It shows up in a bunch of different cancers. And so in the right hand panel, we've got these patients with colon, breast, and lung, and they've all got a little X because that's the BRAF. And the green is some other mutation. And we group them all together and we give them one drug. And Memorial pioneered one of the very first basket trials that David also talked about with the Erdheim-Chester example using vemurafenib FDA approved dose for melanoma V600 mutation BRAF patients but not melanoma, not thyroid, patients who had exhausted therapy looking for signals. And this trial is ongoing and is accumulating patients in different baskets with different diseases who share the BRAF mutation. The one disease, many drugs, many mutations. Again we have all these patients on the left hand side, several different kinds of mutations, three different histologies for simplicity. Now we're going to take all the lung cancer patients on the right side. They've got a bunch of different mutations, in this case three. And the example of this is a trial that has not yet been rolled out but is going to be. And these are the so-called master protocols or platform trials where you have multiple study arms looking at multiple different mutations accruing in parallel allowing you to study these rare genotypes evolving as Dr. Solid said from the battle study experience. So this is the master protocol. We're just going to look at squamous cell lung, 1,000 patients a year, 500 study sites using drugs that have some plausible biological activity. You could do combinations. You can develop them with a diagnostic. And if you hit certain benchmarks like a high response rate, you might even get FDA approval. And this is what it looks like with the master protocol. This is just an over simplified version of it, but you're looking for different biomarkers, A, B, C, and D. You're going to test each of them individually, perhaps against a comparator or maybe not, in this case chemotherapy. And if you don't have any idea what to do at the bottom because you don't have a biomarker, you could always do what's popular today, and that is give them PD-1 directed therapy. And then there's many diseases, many drugs, many mutations. Multiple-biomarker signal finding design, that sounds very nice. One protocol, multiple single arms, non-randomized, and this is what it looks like. So we've got our biomarker profiling. We try to find stuff. If we don't, they go off study. And if we do find stuff, that can go on this massive signal finding trial using biomarker A, B, C, D, E, F, et cetera. On the right hand column you can plug in the biomarker of choice, and what you're doing is conducting multiple parallel Phase 2 trials single arm. And of course we haven't yet agreed among ourselves what is the definition of success. How will we know we're ever done? And also, we are making some assumptions here, which is that if a given set of histologies, 19th-century definitions of cancer, have one mutation that they really are the same disease. But we know that they're really not the same disease. So we bunch them all together, but they're all going to behave differently. It's going to get to be very confusing. So this is the MATCH trial, Molecular Analysis for Therapy Choice. It's a heroic effort, collaboration with multiple drug companies, screening thousands of patients to get 1,000 patients enrolled, again with multiple diseases, multiple mutations, multiple drugs and combinations. And these are the usual eligibility criteria. So, I have some thoughts about what we're going to do about this. I think signal finding, which is where we are in 2014, is not only easy, but it's really cool and a lot of fun. And that's what we're doing right now is we are discovering signals for these drugs and mutations that are very illuminating and incredibly exciting and lead to the types of spectacular anecdotes that Dr. Solid told you about. The hard part comes with validating the positive signals. What the large single arm registration studies? Rituximab, imatinib, ibrutinib, is the proverbial low-hanging fruit. I don't think it's going to be that easy for all of these other drugs and mutations. And I really am worried about randomized trials versus standard of care, because I already told you about a tremendous reluctance to enroll patients or family members to such studies. And for rare mutations, how many centers actually need to participate? And I'm asking you in the audience, could you, could your practice participate financially or logistically if you could only contribute one eligible patient per year? So I think there is an approach, and it has been given different names in the literature, and people are talking about it. But I think a registry approach may be one avenue that's relatively inexpensive. So case reports of off-label uses for rare mutations are biased and not very helpful. But I think if we had national or international registries, perhaps modeled on that registry that all of us participated in for PET scans that was funded by Medicare, we could agree as a community to collect response data in a centralized, unbiased format. And in exchange for being willing to contribute the data-- really simple stuff as David said-- did the patient respond? Did it make any difference to the patient or to you as their doctor? We, the United States of America, we the people, would agree to support or reimburse off-label use. We could create links to some of these new cancer databases where we're going to try to learn from each other and what our practices are in fact doing in real life. And of course it goes without saying that we continue the work pioneered at Memorial of studying the extraordinary responders. I think it's also time for cancer to grow up as a field, and so now that we have pills and we have pills that are relatively nontoxic, it's time for large simple trials. Really folks, the time has come. Broad eligibility criteria, simple enrollment procedures, minimal data collection, endpoints that matter, overall survival, maybe PFS, central IRBs, virtual start-up 100% data capture, trial opens when patients are available, and we ensure that there are adequate per-patient payments. Now, I can think easily of at least 100 reasons why this would not work. But I can think of 101 reasons why the current system definitely does not work. Thank you.